
Premières analyses de la proposition de règlement, publiée par la Commission européenne a publié, le 16 décembre, visant à simplifier et à réviser en profondeur les règlements 2017/745 (MDR) et 2017/746 (IVDR).
La Commission européenne a publié, le 16 décembre, une proposition de règlement visant à simplifier et à réviser en profondeur les règlements (UE) 2017/745 (MDR) et (UE) 2017/746 (IVDR). Cette initiative constitue une étape majeure dans l’évolution du cadre réglementaire européen applicable aux dispositifs médicaux (DM) et aux dispositifs médicaux de diagnostic in vitro (DMDIV).
Deux documents principaux ont été publiés :
- Proposition de règlement modifiant les règlements (UE) 2017/745 et (UE) 2017/746, structurée comme suit :
- Une note explicative (pages 1 à 20, environ 20 pages) ;
- La proposition de règlement modificatif, articulée en cinq articles :
- Article 1 : Modifications du règlement (UE) 2017/745 – MDR (pages 34 à 90, environ 56 pages) ;
- Article 2 : Modifications du règlement (UE) 2017/746 – IVDR (pages 90 à 126, environ 36 pages) ;
- Article 3 : Modifications du règlement (UE) 2022/123 (pages 126 à 127) ;
- Article 4 : Modifications du règlement (UE) 2024/1689 (page 127) ;
- Article 5 : Entrée en vigueur (pages 127 à 128) ;
- Une fiche financière et numérique législative
- Une annexe dédiée aux annexes du MDR et de l’IVDR, détaillant l’ensemble des modifications techniques envisagées, pour un volume d’environ 46 pages.
Les exigences générales de sécurité et de performance (GSPR) ne font pas l’objet de modifications majeures, à l’exception notable :
- des exigences relatives à la suffisance des données cliniques,
- et de la définition de l’équivalence biologique et clinique.
Les évolutions les plus significatives concernent avant tout le processus d’obtention et de maintien du marquage CE.
SIMPLIFICATION ET PROPORTIONNALITE
La Commission européenne propose une série de modifications ciblées du Règlement (UE) 2017/745 relatif aux dispositifs médicaux (MDR) et du Règlement (UE) 2017/746 relatif aux dispositifs médicaux de diagnostic in vitro (IVDR).
Le premier axe de ces évolutions repose sur un objectif central : renforcer la simplification et la proportionnalité du cadre réglementaire, tout en maintenant un haut niveau de sécurité et de performance des dispositifs.
Personne responsable de la conformité réglementaire (PRRC)
MDR : article 15 – IVDR : article 15
La proposition prévoit une suppression des exigences détaillées de qualification actuellement imposées à la PRRC.
Par ailleurs, pour les PME faisant appel à une PRRC externe, l’obligation selon laquelle cette personne doit être « disponible de manière permanente et continue » est supprimée.
Désormais, il sera uniquement exigé que la PRRC soit disponible, ce qui introduit davantage de flexibilité organisationnelle, en particulier pour les structures de taille réduite.
Validité des certificats et recertification
MDR : article 56 – IVDR : article 51
La durée maximale de validité des certificats CE, actuellement fixée à cinq ans, serait supprimée.
En lieu et place d’une recertification formelle, les organismes notifiés mettraient en œuvre des revues périodiques, dont la fréquence et la profondeur seraient proportionnées au niveau de risque du dispositif, tant que le certificat reste valide.
Cette évolution vise à alléger la charge administrative tout en conservant un niveau de surveillance adapté.
Données cliniques, données non cliniques et preuves cliniques
MDR : article 2 point 48, article 61, annexes II et XIV – IVDR : annexe XIII
La proposition élargit la définition des données pouvant être qualifiées de données cliniques, permettant l’utilisation d’un éventail plus large de sources.

Les conditions permettant de s’appuyer sur les données cliniques d’un dispositif équivalent sont assouplies.
Dans l’article 61 du MDR, la possibilité de démontrer la sécurité et la performance d’un dispositif sur la seule base de données non cliniques est étendue.
Par ailleurs, l’utilisation de méthodologies alternatives (« New Approach Methodologies »), telles que les essais in silico, est explicitement encouragée, traduisant une volonté de modernisation des approches d’évaluation.
Technologies bien établies (Well-Established Technologies – WET)
MDR : article 2 point 72, articles 18, 32, 52, 61 et 86
Une nouvelle définition du dispositif à technologie bien établie est introduite.

Ces dispositifs seraient soumis à des exigences plus proportionnées, en remplacement des listes de dispositifs actuellement prévues aux articles 18(3), 52(4) et 61(6)(b) du MDR.
Cette évolution vise à faciliter la conformité des dispositifs disposant d’un historique d’utilisation avéré et d’un profil de sécurité bien documenté.
Reconditionnement et réétiquetage
MDR : article 16 – IVDR : article 16
Les exigences relatives :
- à l’obtention d’un certificat d’organisme notifié,
- ainsi qu’à l’obligation de notification préalable,
pour les activités de reconditionnement et de réétiquetage sont supprimées.

Cette mesure constitue une simplification significative pour les opérateurs concernés, sans remettre en cause les exigences essentielles de sécurité.
Règles de classification
MDR : annexe VIII
Certaines règles de classification sont révisées, entraînant une reclassification vers des classes de risque inférieures pour certains dispositifs, notamment :
- les instruments chirurgicaux réutilisables,
- les accessoires de dispositifs implantables actifs,
- certains logiciels.
Cette adaptation vise à mieux aligner le niveau de contrôle réglementaire avec le risque réel associé à ces dispositifs.
REDUCTION DE LA CHARGE ADMINISTRATIVE
Le deuxième axe des modifications proposées du MDR et de l’IVDR vise explicitement à réduire la charge administrative pesant sur les fabricants, tout en maintenant un niveau de surveillance proportionné au risque et cohérent avec les activités des organismes notifiés.
Résumé de la sécurité et des performances (cliniques) – SS(C)P
MDR : article 32 – IVDR : article 29
Le périmètre des dispositifs pour lesquels le fabricant est tenu de fournir un résumé de la sécurité et des performances (cliniques) est restreint.
Cette obligation ne s’appliquerait désormais qu’aux dispositifs pour lesquels l’organisme notifié doit réaliser une évaluation de la documentation technique.
Dans la mesure où le projet de SS(C)P fait partie intégrante de la documentation soumise à l’organisme notifié, la validation distincte du SS(C)P par l’organisme notifié serait supprimée, simplifiant ainsi le processus d’évaluation.
Rapport périodique actualisé de sécurité (PSUR)
MDR : article 86 – IVDR : article 81
La fréquence de mise à jour obligatoire des rapports périodiques actualisés de sécurité (PSUR) par les fabricants est réduite.
L’examen du PSUR par l’organisme notifié serait intégré aux activités de surveillance menées par celui-ci, plutôt que traité comme une activité indépendante.
Cette évolution vise à rationaliser les interactions entre fabricants et organismes notifiés.
Délais de déclaration de certains incidents graves dans le cadre de la vigilance
MDR : article 87 – IVDR : article 82
Le délai accordé aux fabricants pour déclarer certains incidents graves est allongé.
Pour les incidents ne présentant pas de menace pour la santé publique, et n’entraînant ni décès ni détérioration grave de l’état de santé, le délai de notification passe de 15 à 30 jours.

Cette mesure introduit une plus grande proportionnalité dans le système de vigilance.
Modifications post-certification
MDR : annexe VII – IVDR : annexe VII
Les organismes notifiés devront désormais distinguer clairement :
- les modifications du système de management de la qualité ou du dispositif approuvé pouvant être mises en œuvre sans notification préalable,
- celles pouvant être mises en œuvre sans approbation préalable,
- et celles nécessitant une approbation préalable de l’organisme notifié.
Lorsque cela est pertinent, l’organisme notifié et le fabricant devront convenir d’un plan de gestion des modifications prédéfini (predetermined change control plan), renforçant la prévisibilité et la sécurité réglementaire.
Autorisation ou notification de certaines études de performances
IVDR : article 58
Les études de performances impliquant uniquement des prélèvements sanguins de routine ne seraient plus soumises à une autorisation préalable.
Par ailleurs, l’obligation de notification des études de performances relatives aux diagnostics compagnons, lorsqu’elles utilisent exclusivement des échantillons résiduels (leftover specimens), serait supprimée.

Ces ajustements visent à faciliter la conduite d’études à faible risque, tout en préservant l’évaluation éthique et scientifique appropriée.
INNOVATION ET DISPONIBILITE DES DISPOSITIFS POUR DES CAS SPECIFIQUES
Le troisième axe des modifications proposées du MDR et de l’IVDR vise à favoriser l’innovation, à sécuriser l’accès aux dispositifs pour des populations spécifiques ou dans des contextes particuliers, et à renforcer la capacité de réponse du cadre réglementaire européen face aux évolutions technologiques et aux situations de crise.
Dispositifs « in house »
MDR : article 5(5) – IVDR : article 5(5)
Les conditions encadrant la fabrication et l’utilisation de dispositifs au sein des établissements de santé sont assouplies.
Il serait notamment possible de transférer des dispositifs in house lorsque cela est justifié par l’intérêt de la sécurité des patients ou de la santé publique.
Sous l’IVDR, la condition selon laquelle aucun dispositif équivalent ne doit être disponible sur le marché est supprimée.
Par ailleurs, les laboratoires centraux fabriquant et utilisant des tests exclusivement dans le cadre d’essais cliniques sont explicitement intégrés au champ de l’exemption applicable aux dispositifs in house.
Interruption ou arrêt de mise à disposition de certains dispositifs
MDR : article 10a – IVDR : article 10a
Un outil informatique centralisé destiné au signalement et à l’échange d’informations sera mis en place dans Eudamed, ou via des systèmes interopérables avec celui-ci.
L’Agence européenne des médicaments (EMA) sera chargée de :
- développer une méthodologie d’identification des dispositifs soumis à l’obligation de déclaration,
- établir et maintenir une liste des dispositifs concernés.
Cette mesure vise à anticiper et à limiter les ruptures d’approvisionnement critiques.
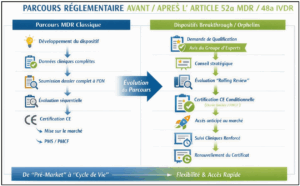
Procédures d’évaluation de la conformité pour les dispositifs innovants et orphelins
MDR : nouvel article 52a – IVDR : nouvel article 48a
L’introduction du nouvel article « Conformity assessment of breakthrough devices and of orphan devicess » vise à corriger certains effets non intentionnels du cadre actuel, notamment :
- les délais excessifs d’accès au marché ;
- la difficulté d’appliquer de manière strictement uniforme les exigences de preuves cliniques à des dispositifs pour lesquels la génération de données cliniques robustes avant mise sur le marché est structurellement complexe ;
- la perte d’attractivité de l’Union européenne par rapport à d’autres juridictions (FDA Breakthrough Devices Program, orphan pathways).
L’article 52a/48a introduit ainsi une voie réglementaire dédiée avec des critères spécifiques pour la qualification des dispositifs innovants (« breakthrough devices ») et des dispositifs orphelins.
Après une Qualification par un panel d’experts, ces dispositifs bénéficieront :
- d’une évaluation prioritaire,
- d’un examen progressif (« rolling review »),
- et d’un accès structuré aux avis des panels d’experts.
L’objectif est de faciliter et d’accélérer l’accès au marché européen pour des dispositifs répondant à des besoins médicaux non couverts.
| Dispositifs de rupture (breakthrough devices) | Dispositifs orphelins |
|
Le texte introduit pour la première fois dans le MDR une notion proche du concept de breakthrough innovation déjà reconnu au niveau international. Trois éléments clés caractérisent cette catégorie :
Cette définition crée une rupture conceptuelle avec l’approche purement fondée sur la classification de risque (classes I, IIa, IIb, III), en intégrant une dimension de valeur clinique et de besoin médical non satisfait. |
Le MDR reconnaît désormais explicitement une catégorie équivalente aux dispositifs destinés aux maladies rares, avec :
Cette reconnaissance constitue un alignement partiel avec les logiques déjà en place pour les médicaments orphelins, tout en tenant compte des spécificités des dispositifs médicaux. |
L’alinéa 7 introduit une logique proche de la mise sur le marché conditionnelle, reposant sur :
- un rapport bénéfice-risque favorable ;
- un engagement fort du fabricant en post-market clinical follow-up (PMCF) ;
- la possibilité de certificats à durée limitée assortis de conditions spécifiques.
Dérogations en cas d’urgence de santé publique, de catastrophe ou de crise
MDR : article 59 et nouvel article 59a – IVDR : article 54 et nouvel article 54a
La Commission européenne pourra, de sa propre initiative, autoriser la mise sur le marché de dispositifs en cas d’urgence de santé publique.
Les autorités compétentes nationales pourront, quant à elles, autoriser des dérogations aux exigences de fabrication, de conception ou de destination de dispositifs déjà marqués CE, en cas de menaces transfrontalières graves pour la santé, de catastrophes ou de crises majeures.
Espaces d’expérimentation réglementaire (« regulatory sandboxes »)
MDR : nouveaux articles 59b et 59c – IVDR : nouveaux articles 54b et 54c
Les États membres et la Commission pourront mettre en place des espaces d’expérimentation réglementaire, afin de répondre aux besoins spécifiques liés aux technologies émergentes.
Ces dispositifs visent à permettre le test encadré de solutions innovantes dans un environnement réglementaire sécurisé.
Reconditionnement des dispositifs à usage unique
MDR : article 17
Les fabricants devront désormais justifier explicitement l’allégation « usage unique ».
Tous les dispositifs qui ne sont pas destinés à un usage unique pourront être retraités conformément aux instructions fournies par le fabricant.
Toute personne procédant à la remise à neuf complète d’un dispositif à usage unique sera considérée comme le fabricant du dispositif remis à neuf.
Ces dispositions deviendront applicables cinq ans après l’entrée en vigueur du règlement modifié.
Kits de diagnostic
IVDR : nouvel article 19a
Des clarifications sont apportées concernant la composition des kits, tels que définis à l’article 2, point 11, de l’IVDR, afin de sécuriser leur qualification réglementaire.
Jusqu’à présent, la composition des kits soulevait des incertitudes d’interprétation, notamment lorsque ceux-ci incluaient :
- des IVD non marqués CE pris individuellement,
- des dispositifs médicaux relevant du MDR,
- ou des produits ne relevant pas stricto sensu de la réglementation DM/DMDIV.
L’article 19a apporte ainsi une base juridique explicite pour encadrer ces configurations.

Maintien sur le marché des dispositifs orphelins existants (« grandfathering »)
MDR : article 120 – IVDR : article 110
Les dispositifs orphelins ayant obtenu le marquage CE sous les anciennes directives, et pour lesquels un panel d’experts a confirmé le respect des critères de « dispositif orphelin », pourront continuer à être mis sur le marché au-delà des périodes de transition, sous réserve de conditions spécifiques.
Nanomatériaux
MDR : annexes I et VIII
La définition obsolète du nanomatériau figurant à l’article 2 du MDR est supprimée.
Elle est remplacée par une référence à la recommandation de la Commission du 10 juin 2022, laquelle s’appliquera aux dispositions relatives aux nanomatériaux figurant dans les annexes I et VIII.

PREVISIBILITE ET EFFICACITE ECONOMIQUE DE LA CERTIFICATION
Le quatrième axe des modifications proposées du MDR et de l’IVDR vise à renforcer la prévisibilité des processus de certification, à améliorer leur efficience économique, et à optimiser l’allocation des ressources des organismes notifiés, en particulier pour les dispositifs à risque faible ou modéré.
Dialogue structuré entre fabricants et organismes notifiés
MDR : annexe VII – IVDR : annexe VII
Une base juridique explicite est introduite afin de permettre aux fabricants et aux organismes notifiés de mener un dialogue structuré, avant et après la soumission de la documentation de certification.
Ce dialogue devra s’appuyer sur des procédures formalisées et documentées, favorisant une meilleure anticipation des attentes réglementaires et une réduction des itérations inutiles lors de l’évaluation de la conformité.
Procédures d’évaluation de la conformité
MDR : article 52, annexes IX, X et XI – IVDR : article 48, annexes IX, X et XI
L’implication des organismes notifiés dans l’évaluation de la conformité des dispositifs à risque faible et intermédiaire est significativement réduite :
- Pour les dispositifs de classe IIa et IIb (MDR) et B et C (IVDR), l’évaluation de la documentation technique pourra être limitée :
- à un dispositif représentatif pour un groupe générique,
- pour une catégorie de dispositifs,
- ou pour l’ensemble du portefeuille du fabricant.
- Les revues systématiques de la documentation technique de dispositifs représentatifs lors des activités de surveillance seront supprimées.
- Les DMDIV de classe A stériles ne nécessiteront plus l’intervention d’un organisme notifié.
En parallèle :
- les audits sur site pourront être remplacés par des audits à distance, lorsque cela est justifié,
- les audits de surveillance pourront être réalisés tous les deux ans en l’absence de problématiques de sécurité,
- les audits inopinés seront limités aux situations justifiées (« for cause »).
Les délais de consultation des autorités compétentes pour les médicaments et les substances d’origine humaine (SoHO) seront également réduits.
Procédures de consultation clinique et de performance
MDR : article 54 – IVDR : article 48 et nouvel article 56a
Le périmètre de la procédure de consultation de l’évaluation clinique (CECP) est restreint aux dispositifs implantables de classe III.
La Commission se voit toutefois conférer la possibilité d’étendre ce périmètre à d’autres types de dispositifs par acte délégué.
Pour les DMDIV, la procédure de consultation de l’évaluation des performances (PECP) est supprimée.
En contrepartie, une possibilité d’avis précoce des panels d’experts est introduite pour les dispositifs de classe C et D, favorisant une anticipation des exigences scientifiques et réglementaires.
Frais des organismes notifiés
MDR : article 50
Des réductions de frais sont prévues :
- pour les micro-entreprises et petites entreprises,
- ainsi que pour les dispositifs orphelins.
La Commission européenne sera habilitée à définir le niveau et la structure des frais facturés par les organismes notifiés, afin d’harmoniser les pratiques et d’améliorer la transparence des coûts de certification au niveau européen.
D’autre part : Vers une pratique harmonisée et des délais prévisibles pour les organismes notifiés
Avant même la publication de la proposition, le 12 décembre 2025, la Commission européenne a publié son projet de règlement d’exécution relatif aux exigences de qualité et aux procédures des activités de certification des organismes notifiés. Bien que ce texte ne fasse pas formellement partie de l’initiative de simplification du MDR et de l’IVDR, il s’inscrit dans l’objectif global de la Commission : offrir un cadre réglementaire plus prévisible pour l’industrie MedTech.
Le projet de règlement d’exécution prévoit notamment :
- de préciser et limiter les informations que les organismes notifiés doivent demander avant de fournir un devis, ainsi que le contenu minimum de ce devis ;
- de fixer des délais maximaux pour les activités d’évaluation de la conformité (255 jours au total, ou 165 jours si le QMS et la vérification produit peuvent être réalisés en parallèle) et pour l’évaluation des modifications substantielles planifiées (135 jours, sauf si une nouvelle évaluation de conformité est nécessaire) ;
- de définir les règles et durées maximales de suspension des activités d’évaluation de la conformité lorsque des informations supplémentaires sont requises auprès du fabricant ;
- de préciser la documentation que les organismes notifiés doivent demander pour la recertification, avec un délai maximal de 60 jours pour l’examen de cette documentation ;
- de fixer un délai maximal de 60 jours pour vérifier les demandes de recertification du système de management de la qualité (QMS) ;
- de prévoir un délai maximal de 15 jours pour la réémission des certificats après décision positive.
La Commission prévoit d’adopter ce règlement d’exécution au premier trimestre 2026, avec une application progressive : certaines dispositions dès trois mois après l’entrée en vigueur, et d’autres au plus tard le 1er janvier 2028. La Commission accepte les retours des parties prenantes sur ce projet de règlement jusqu’au 23 janvier 2026.
RENFORCEMENT DE LA COORDINATION AU SEIN DU SYSTEME DECENTRALISE
Le cinquième axe des modifications proposées du MDR et de l’IVDR vise à améliorer la cohérence, la transparence et l’efficacité du système réglementaire européen, fondé sur une gouvernance décentralisée reposant sur les autorités compétentes nationales, les organismes notifiés et les instances européennes de coordination.
Statut réglementaire des produits et classification des dispositifs
MDR : article 4, nouveaux articles 4a, 51a et 51b – IVDR : article 3, nouveaux articles 3a, 47a et 47b
La coordination entre autorités compétentes concernant :
- la qualification réglementaire d’un produit,
- et la classification d’un dispositif,
est formalisée et codifiée, sur la base de la procédure dite « procédure d’Helsinki ».
Cette procédure permettra, le cas échéant, de solliciter l’avis des panels d’experts, afin d’harmoniser les décisions au niveau européen et de réduire les divergences d’interprétation entre États membres.
Mécanisme de résolution des différends entre fabricants et organismes notifiés
MDR : article 35 – IVDR : article 31
L’autorité compétente en charge des organismes notifiés se verra attribuer un rôle de médiateur (« ombudsman ») en cas de litige entre un fabricant et un organisme notifié.
Ce mécanisme vise à renforcer la sécurité juridique et à prévenir les blocages prolongés dans les processus de certification.
Renforcement du rôle de l’expertise externe dans le système réglementaire
MDR : article 106 et nouvel article 106a – IVDR : article 100
Le rôle des panels d’experts est élargi, tant en termes de missions que de composition.
Ils pourront notamment intervenir dans :
- la détermination du statut réglementaire des produits,
- la classification des dispositifs.
Les panels d’experts seront en mesure de fournir des avis scientifiques, techniques, cliniques et réglementaires à la Commission européenne, aux États membres, au MDCG, aux organismes notifiés et, dans certains cas, aux fabricants.
L’Agence européenne des médicaments (EMA) continuera d’assurer le secrétariat des panels d’experts.
Les fonctions respectives des panels d’experts et des laboratoires d’experts, actuellement regroupées au sein de l’article 106 du MDR, seront clarifiées par l’introduction d’une disposition spécifique dédiée aux laboratoires d’experts.
Appui de l’EMA à la coordination des autorités compétentes
MDR : nouvel article 106b
L’EMA apportera un soutien scientifique, technique et administratif à la coordination entre autorités compétentes nationales, notamment dans les domaines suivants :
- produits frontières et classification,
- études cliniques multinationales,
- dérogations,
- vigilance,
- surveillance du marché.
L’EMA fournira également un soutien spécifique aux PME, afin de faciliter leur navigation dans le système réglementaire européen.
POURSUITE DE LA DIGITALISATION
Le sixième axe des modifications proposées du MDR et de l’IVDR s’inscrit dans une dynamique de digitalisation accrue des processus de conformité, de certification et de mise sur le marché, avec pour objectif d’améliorer l’efficacité opérationnelle, la traçabilité et l’accessibilité de l’information pour l’ensemble des acteurs du système.
Digitalisation des outils de conformité
MDR : article 19, nouvel article 110a, annexes I et VI – IVDR : article 17, nouvel article 103a, annexes I et VI
Plusieurs évolutions majeures sont introduites :
- La déclaration UE de conformité pourra être fournie sous forme numérique.
- Sous réserve de futures règles d’exécution, certaines informations figurant sur l’étiquetage pourront être communiquées sous forme numérique.
- Les fabricants de tests destinés à être utilisés à proximité du patient (near patient tests) pourront fournir des instructions d’utilisation électroniques.
- La transmission des informations exigées par le MDR et l’IVDR devra être réalisée par voie électronique.
- Les opérateurs économiques devront renseigner leurs coordonnées numériques dans Eudamed.
Ces mesures visent à moderniser les supports de conformité tout en renforçant leur accessibilité.
Digitalisation de l’évaluation de la conformité
MDR : nouvel article 52b – IVDR : nouvel article 48b
Les fabricants pourront établir :
- la documentation technique,
- les rapports d’évaluation,
- et les autres documents réglementaires,
exclusivement sous forme numérique, ce qui devrait faciliter les échanges avec les organismes notifiés et réduire les délais de traitement.
Vente en ligne de dispositifs
MDR : article 6 – IVDR : article 6
Dans le cadre des ventes en ligne, certaines informations essentielles permettant d’identifier le dispositif, ainsi que les instructions d’utilisation, devront être mises à disposition de l’utilisateur final.
Cette disposition vise à garantir un niveau d’information équivalent à celui disponible lors de la mise sur le marché par des canaux traditionnels.
Identifiant unique du dispositif (UDI) et Eudamed
MDR : articles 27 et 33, annexe VII – IVDR : articles 24 et 30, annexe VII
Les dispositions relatives à :
- l’attribution de l’UDI,
- et à son enregistrement dans Eudamed,
sont clarifiées afin d’améliorer la cohérence et l’interopérabilité du système.
Il est également prévu que certains systèmes électroniques puissent être mis en place en dehors d’Eudamed, sous réserve d’interopérabilité, offrant ainsi une flexibilité accrue dans l’architecture numérique européenne.
Coopération internationale
Le septième axe des modifications proposées du MDR et de l’IVDR introduit un cadre formalisé de coopération réglementaire internationale, visant à renforcer la convergence des exigences, à favoriser la reconnaissance mutuelle et à améliorer l’efficacité globale des systèmes de réglementation des dispositifs médicaux.
MDR : nouveaux articles 108a et 108b
Une nouvelle section dédiée à la coopération internationale est introduite dans le MDR.
Elle vise à promouvoir des initiatives favorisant la convergence réglementaire mondiale et le renforcement de la coopération entre autorités compétentes.
Les activités explicitement soutenues incluent notamment :
- la participation active aux travaux du International Medical Device Regulators Forum (IMDRF),
- l’appui aux mécanismes de reconnaissance et de confiance mutuelle, tels que le Medical Device Single Audit Program (MDSAP).
L’introduction de ces dispositions ouvre la voie à une utilisation accrue des mécanismes de reliance, permettant de s’appuyer, lorsque cela est pertinent, sur les évaluations réalisées par des autorités ou des systèmes réglementaires tiers reconnus.
Ce volet marque une étape importante vers une approche plus intégrée et plus internationale de la réglementation des dispositifs médicaux, susceptible de réduire les redondances d’évaluation et de faciliter l’accès des dispositifs au marché mondial.
Articulation avec les autres législations de l’Union européenne
Le huitième axe des modifications proposées du MDR et de l’IVDR vise à renforcer la cohérence du cadre réglementaire européen, en assurant une meilleure articulation entre la réglementation des dispositifs médicaux et d’autres textes législatifs de l’Union, notamment dans les domaines des essais cliniques et de la cybersécurité.
Études combinées impliquant des médicaments, des dispositifs médicaux et/ou des DMDIV
MDR : nouvel article 79a – IVDR : nouvel article 75a
Pour les études combinées impliquant :
- un médicament et un dispositif médical,
- un médicament et un dispositif médical de diagnostic in vitro,
le promoteur pourra soumettre une demande unique, déclenchant une évaluation coordonnée.
Cette évaluation sera réalisée conformément au règlement (UE) n° 536/2014 relatif aux essais cliniques, lequel sera modifié en conséquence par le Biotech Act afin d’intégrer ce nouveau dispositif.
Cette approche vise à réduire la fragmentation réglementaire et à simplifier la conduite d’études multidisciplinaires au sein de l’Union européenne.
C’est ainsi l’occasion d’introduire une nouvelle définition « combined study » à l’ article 2 :

Cybersécurité
MDR : nouvel article 87a et annexe I – IVDR : nouvel article 82a et annexe I
Les incidents graves déclarés dans le cadre du système de vigilance du MDR ou de l’IVDR, et qui correspondent également à des :
- vulnérabilités activement exploitées,
- ou à des incidents graves au sens du règlement (UE) 2024/2847 relatif à la cyberrésilience,
seront transmis aux :
- équipes nationales de réponse aux incidents de sécurité informatique (CSIRTs),
- ainsi qu’à l’Agence de l’Union européenne pour la cybersécurité (ENISA).
En complément, les fabricants devront également déclarer via Eudamed les vulnérabilités activement exploitées et les incidents graves relevant de la cybersécurité qui ne constituent pas des incidents graves au sens du MDR ou de l’IVDR, à destination des CSIRTs et de l’ENISA.
Enfin, la cybersécurité est explicitement intégrée aux exigences générales de sécurité et de performance figurant à l’annexe I du MDR et de l’IVDR, renforçant ainsi son statut en tant qu’exigence réglementaire à part entière.
WHAT’S NEXT
Et après ?
La proposition législative visant à simplifier les règlements MDR et IVDR sera désormais soumise au Parlement européen et au Conseil de l’Union européenne pour adoption.
Une fois adoptées par les co-législateurs, la plupart des simplifications prévues s’appliqueront six mois après l’entrée en vigueur et certaine jusqu’à 5 ans après.
La Commission s’attend à une adoption par les colégislateurs (Parlement européen et Conseil de l’UE) d’ici le deuxième trimestre 2027 (Q2 2027), avec une applicabilité potentielle à la même période. Cela correspond à un processus législatif d’environ 18 mois, ce qui est réaliste pour une proposition complexe mais prioritaire, sans urgence extrême.
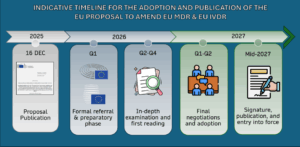
Voici le calendrier prévisionnel ajusté en intégrant cette attente officielle :
| Période prévisionnelle | Étape |
| 16 décembre 2025 | Publication et adoption par la Commission européenne |
| Janvier – mars 2026 | Saisine officielle et préparation |
| Avril – décembre 2026 | Examen en profondeur et première lecture |
| Janvier – juin 2027 (Q1-Q2 2027) | Négociations finales et adoption |
| Mi-2027 (probable) | Signature, publication et entrée en vigueur |
Notes importantes :
Ce calendrier est plus court que la moyenne (18-24 mois) en raison de la priorité accordée par la Commission et les parties prenantes. Le processus pourrait s’accélérer avec un accord politique fort, ou s’allonger en cas de divergences majeures.
Ainsi, si la proposition de la Commission constitue un jalon important vers un cadre réglementaire plus favorable à l’industrie MedTech en Europe, l’industrie devra encore patienter avant de bénéficier concrètement d’un allègement des règles actuellement complexes.
Dans l’intervalle, nous continuerons à suivre de près l’évolution de cette proposition, et à rapporter les étapes clés.
Source 1 : Commission Européenne
Source 2 : Commission Européenne
